Primary Focus
RPE mitochondrial dysfunction is thought to play a causative role in retinal degenerative diseases such as mitochondrial retinopathy and age-related macular degeneration. As a test of this hypothesis, we generated mice with an RPE-selective postnatal loss of mitochondrial oxidative phosphorylation (OXPHOS). OXPHOS-deficient RPE cells are surprisingly long-lived, but lose critical epithelial characteristics through cellular dedifferentiation and, later, an epithelial to mesenchymal-like transition. OXPHOS-deficient RPE cells initiate a stress response that includes dependence upon the HGF/c-Met pathway, upregulation of aerobic glycolysis, activation of the mTOR signaling pathway, and cellular hypertrophy. Activation of mTOR and subsequent dedifferentiation can also be triggered by acute chemical oxidative damage to the RPE in vivo. For both chronic metabolic and acute oxidative RPE stress, the consequences for adjacent photoreceptors are profoundly negative, resulting in a gradual or rapid (respectively) retinal degeneration. Strikingly, treatment of animals with the mTOR inhibitor, rapamycin, blunts RPE dedifferentiation and hypertrophy and preserves photoreceptor numbers and function for both stressors. We would like to understand the mechanism of mTOR-mediated RPE dedifferentiation and determine whether this new in vivo RPE stress response is activated in human retinal disease.
Secondary Focus
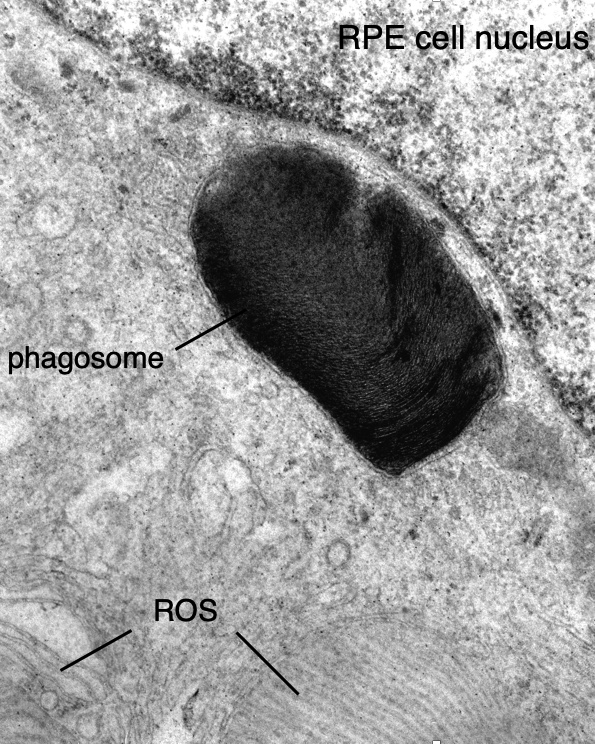
Phagocytosis is an example of a basic process that we study. Every morning in mammalian eyes, the distal portion of the light sensing outer segments of photoreceptors are phagocytized by adjacent cells of the retinal pigment epithelium (RPE). Phagocytosis is balanced by new synthesis at the proximal end of the outer segment. Together, these two processes lead to constant turnover of outer segments and serve to repair light- and oxygen-induced damage. The daily “big breakfast” of outer segment material, summed over the life of an animal, distinguishes the post-mitotic RPE cell as the most phagocytic cell in the body. Photoreceptor degeneration in mutant rats and mice with defective RPE phagocytosis demonstrates that this process is essential for the normal functioning of the mammalian retina. By genetic analysis of these mutant rodents, we identified the receptor tyrosine kinase, MERTK, as a critical part of the phagocytic mechanism. We also identified mutations in the human MERTK gene in individuals with a retinal degenerative disease known as retinitis pigmentosa. We have elaborated our understanding of the mechanism of phagocytosis by demonstrating that MERTK acts locally, at the site of phagocytosis, to promote ingestion of bound outer segment tips. MERTK does so by triggering a striking redistribution of myosin II from the cell periphery to sites of ingestion. We are continuing to investigate the mechanism of RPE phagocytosis with an emphasis on identifying new protein components and understanding its circadian regulation.